Types of Wolfram Syndrome
Understanding the genetic distinctions and physiological impacts of the different forms of Wolfram Syndrome.
Type 1 (WFS1)
Type 1 is the most common form of Wolfram Syndrome. It is an autosomal recessive disorder caused by mutations in the WFS1 gene, which provides instructions for producing a protein called wolframin.
Wolframin is primarily located in the endoplasmic reticulum (ER) of cells, where it plays a critical role in regulating calcium levels and managing protein folding. When wolframin is defective or absent, cells experience ER stress, leading to cellular dysfunction and premature cell death, particularly in the pancreas and nervous system.
This type typically presents with the classic symptoms: early-onset Diabetes Mellitus, Optic Atrophy, Deafness, and Diabetes Insipidus.
Type 2 (CISD2)
Type 2 is a rarer variant of Wolfram Syndrome. It is caused by mutations in the CISD2 gene.
The CISD2 gene provides instructions for making a protein that is located in the outer membrane of mitochondria, the energy-producing centers of cells. Mutations in this gene disrupt mitochondrial function and cellular energy production.
While Type 2 shares many symptoms with Type 1 (such as diabetes mellitus and optic atrophy), it is uniquely associated with defective blood clotting (bleeding tendency) and gastrointestinal ulcers. Patients with Type 2 typically do not develop diabetes insipidus.
WFS1-Related Disorder
Individuals with dominant, pathogenic WFS1 mutations have WFS1-related disorder.
Unlike the classic autosomal recessive Type 1, these single mutations of the WFS1 gene are present in a distinct subset of patients. They often exhibit some, but not all, of the typical symptoms of Wolfram Syndrome.
This spectrum can include isolated hearing loss, cataracts, or adult-onset diabetes. Because the symptoms are variable and often milder, this related disorder can sometimes go unrecognized or be misdiagnosed for years.
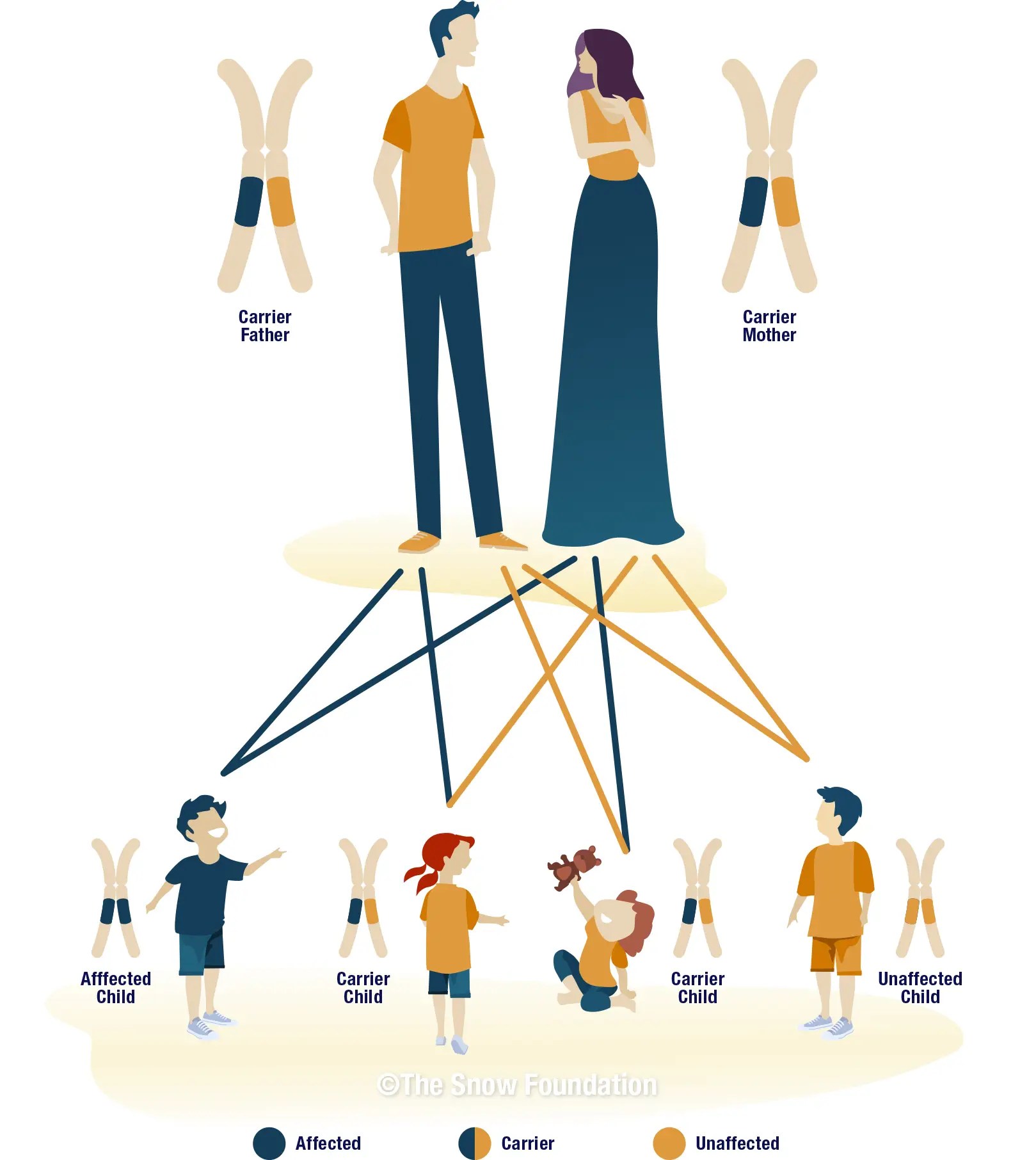
Autosomal Recessive Inheritance
Wolfram Syndrome Type 1 and Type 2 are both autosomal recessive genetic disorders. This means that for a child to be born with the condition, they must inherit two mutated copies of the gene—one from each parent.
Parents of a child with an autosomal recessive condition are typically "carriers." They carry one copy of the mutated gene and one normal copy, but they usually do not show symptoms of the disease themselves.
When two carriers have a child together, there is a:
- 25% chance the child will have Wolfram Syndrome.
- 50% chance the child will be a carrier, like the parents.
- 25% chance the child will neither have the syndrome nor be a carrier.
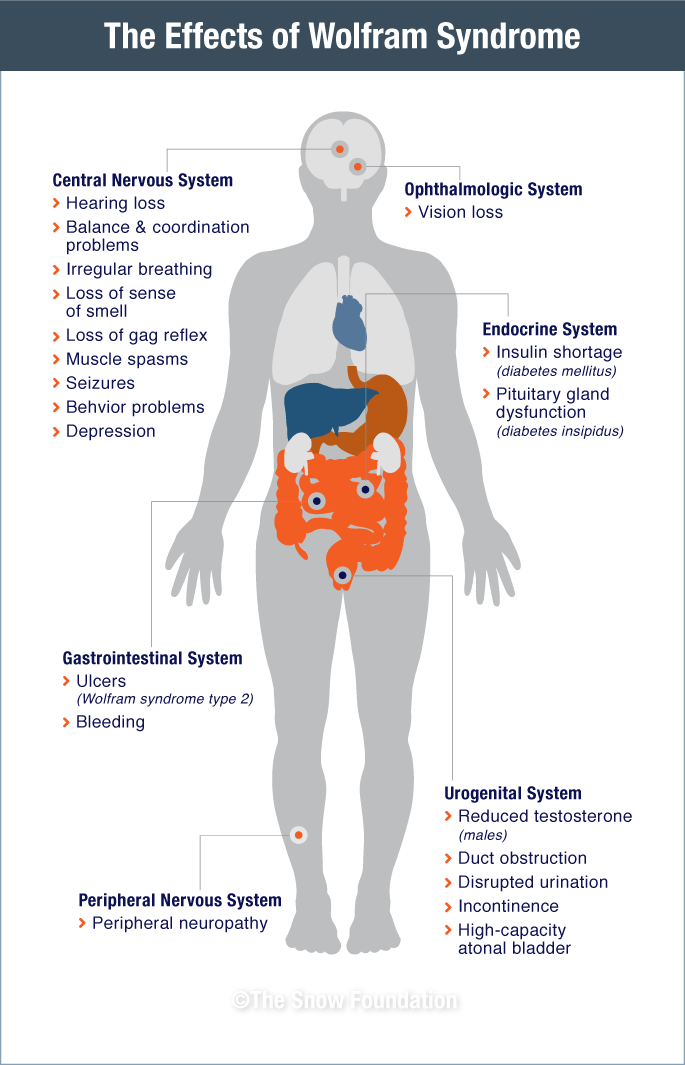
The Systemic Effects
Wolfram Syndrome affects multiple organ systems throughout the body. The diagram below illustrates the widespread physiological impacts of the disease.
Cellular Impact: Normal vs. Wolfram Syndrome
The symptoms of Wolfram Syndrome are caused by a dysfunctional wolframin protein (WFS1), leading to endoplasmic reticulum (ER) stress and ultimately cell death. In normal cells, WFS1 helps regulate ATF6 to restore ER homeostasis — in Wolfram Syndrome, this pathway fails, triggering apoptosis.

Normal Cell Response
When ER stress occurs in a healthy cell, WFS1 is induced and works with HRD1 to degrade ATF6α via the proteasome. This negative feedback loop restores ER homeostasis, allowing the cell to survive and function normally.

Wolfram Syndrome Cell Response
With defective WFS1, the regulatory pathway breaks down. ATF6 activates CHOP, a pro-apoptotic transcription factor, which drives the cell toward apoptosis — resulting in a collapsed nucleus, apoptotic bodies, and ultimately cell death.